INTRODUCTION
Cardiac arrest (CA), also called cardiopulmonary arrest or circulatory arrest, is a sudden cessation in normal blood circulation because the heart has failed to pump blood [1]. Most studies on CA over the past half-century have focused on improving the rate of return of spontaneous circulation (ROSC), and significant progress has been made. However, even though immediate resuscitation can increase ROSC, prognosis remains poor [2-4]. The low survival rate of patients who have ROSC after CA can be attributed to a unique pathophy siological process [1] known as the post CA syndrome (PCAS). In fact, PCAS is recognized as the main cause of low survival rate after ROSC [3]. The early-phase PCAS survival rate in patients is 4% to 33%, depending on the chain of survival [4]. Such a low survival rate in patients with PCAS seems to result from a combination of whole-body ischemia/reperfusion-mediated damage and nonspecific activation of a systemic inflammatory response by CA and ROSC [2].
Many studies have focused on myocardial and brain injury and dysfunction after CA [5,6]. There is no doubt that the heart is important in PCAS. Recently, Roberts et al. [7] reported that multiple organ dysfunction is common after ROSC in patients with PCAS, and is associated with a low survival rate. Nevertheless, there is a paucity of information on inflammation in and histopathology of hearts after CA. Furthermore, the relationship between survival and cardiac damage remains unclear in cases of PCAS.
Systemic ischemia/reperfusion injury after ROSC triggers the release of inflammatory cytokines, leading to a systemic inflammatory response syndrome that mimics sepsis, even in the absence of infection [8]. The inflammatory response after ROSC is characterized by polymorphonuclear leukocyte activation, adhesion molecule expression, reactive oxygen species production in response to inducible nitric oxide synthase, and release of cytokines such as tumor necrosis factor (TNF)-α [9]. However, the mechanisms underlying this response in CA and PCAS remain unclear.
Therefore, the inflammatory response in the heart following CA should be studied, as well as the survival rate in early-phase PCAS. Therefore, we induced asphyxial CA in rats and determined the survival rate during the post resuscitation phase. Moreover, we studied histopathological changes in the heart after ROSC and investigated changes in the levels of the pro-inflammatory cytokine TNF-α after ROSC by immunohistochemistry.
METHODS
Experimental animals and groups
Male Sprague-Dawley rats were obtained from the experimental animal center of Kangwon National University (Chuncheon, South Korea). All experimental protocols were approved based on ethical and scientific guidelines of the University Institutional Animal Care and Use Committee (approval no. KW-151127-1). Experimental animals were divided into the following groups: 1) sham-CA rats (n=7 for each time point examined), which did not undergo CA procedures but otherwise were treated with the same conditions as the CA group; and 2) CA rats (n=7 for each time point examined), which were subjected to CA procedures. The rats in each group were sacrificed at 6, 12, 24 hours, and 2 days after ROSC.
CA induction and cardiopulmonary resuscitation
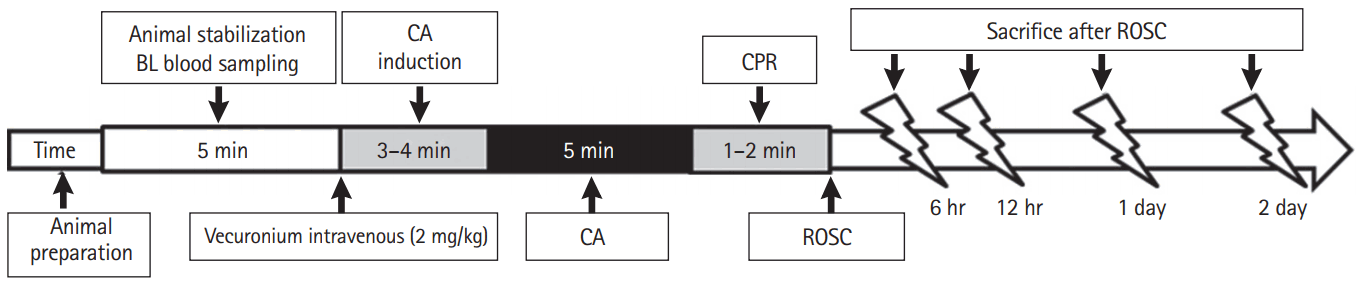
CA and cardiopulmonary resuscitation (CPR) was performed using published procedures [10,11] with minor modifications (Fig. 1). Briefly, rats were anesthetized with 2% to 3% isoflurane and mechanically ventilated using a rodent ventilator (Harvard Apparatus, Holliston, MA, USA) to maintain respiration. To monitor peripheral oxygen saturation (SpO2), an oxygen saturation probe (Nonin Medical, Plymouth, MN, USA) was attached to the left foot of each rat. Body temperature was maintained at 37±0.5°C during and after the CA procedures. To monitor electrical activity, electrocardiographic probes (GE Healthcare, Milwaukee, WI, USA) were placed (three leads per limb), and electrocardiograms were examined continuously. The left femoral artery and right femoral vein were separately cannulated to monitor mean arterial pressure (MAP) (MLT 1050/D; AD Instruments, Bella Vista, Austria) and administer drugs intravenously.
After 5 minutes of stabilization, vecuronium bromide (2 mg/kg; Gensia Sicor Pharmaceuticals, Irvine, CA, USA) was intravenously administered and anesthesia and mechanical ventilation were stopped. A MAP below 25 mmHg and subsequent pulseless electric activity were used to define CA [11,12]. CA was confirmed at 3 to 4 minutes after vecuronium bromide injection. Five minutes after CA, CPR was initiated by intravenously administering a bolus of epinephrine (0.005 mg/kg) and sodium bicarbonate (1 mEq/kg), followed by mechanical ventilation with 100% oxygen and mechanical chest compressions at a rate of 300/min until MAP reached 60 mmHg and electrocardiographic activity was observed. Once the animal was hemodynamically stable and spontaneously breathing, usually at 1 hour after ROSC, 2 hours after resuscitation, and with rats under anesthesia with isoflurane, the arterial and venous catheters were removed, and the wounds were closed. The rats were then mechanically ventilated with ambient air and allowed to extubate themselves. After extubation, the rats were returned to their cages and monitored closely for long-term outcomes. Thereafter, animals were subcutaneously administered 20 mL/kg/day isotonic saline with 5% dextrose until they could eat and drink without assistance. Sham animals received the same treatment except for the induction of CA and administration of CPR.
H&E staining and TNF-α immunohistochemistry
H&E staining and TNF-α immunohistochemistry were performed according to published procedures developed in our laboratory [13]. In short, animals were anesthetized with 30 mg/kg Zoletil 50 (Virbac, Carros, France) and perfused transcardially with 4% paraformaldehyde. Hearts were cut sagittally, embedded in paraffin, and then sectioned (6 µm).
To examine histopathological changes, the sections were stained with H&E, dehydrated in an ethanol series, and mounted with Canada balsam (Kanto Chemical, Tokyo, Japan). The histopathology of heart lesions was semiquantitatively assessed using a published procedure [14]. In brief, we counted cells with eosinophilic cytoplasms, darker nuclei, cytoplasmic vacuolization, infiltration, and congestion. Cells with these effects in lesions were graded using the following scale: 0, none; 1, mild; 2, moderate; 3, severe.
To examine changes in TNF-α levels, tissue sections were incubated with rabbit anti-TNF-α (diluted 1:500; Abcam Incorporated, Cambridge, MA, USA), followed by addition of secondary antibody (Vector Laboratories, Burlingame, CA, USA), and then developed using Vectastain ABC (Vector Laboratories) and visualized using 3,3′-diaminobenzidine.
To quantitatively analyze TNF-α immunoreactivity, five microscope fields (×200 magnification) in the posterior wall of the left ventricle and septum of the heart were randomly selected, and the staining intensities of TNF-α-immunoreactive structures were evaluated based on the optical density (OD) obtained after transformation of the mean level of gray using the formula: OD=log (256/mean gray level). The OD of the background was establishing using areas adjacent to the evaluated area. After the background density was subtracted, ratios of ODs in the image files were calibrated in Photoshop ver. 8.0 (Adobe Systems, San Jose, CA, USA), and presented as percentages, with ODs of stained tissues from rats in the sham-CA group set at 100% in NIH Image ver. 1.59 (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data were entered into SAS ver. 9.02 (SAS Institute, Cary, NC, USA) and presented as means±standard error of the mean (SEM). Survival was analyzed using the Kaplan-Meier statistic and log-rank test. MAP and peripheral oxygen levels were compared by one- and two-way repeated measures ANOVAs to assess the effects of time. To determine the significance of differences, post hoc Tukey testing was conducted for all pairwise multiple comparisons. For semiquantitative analyses of histopathology and TNF-α immunoreactivity, differences were considered significant when P-values were less than 0.05.
RESULTS
Physiological variables and survival rate
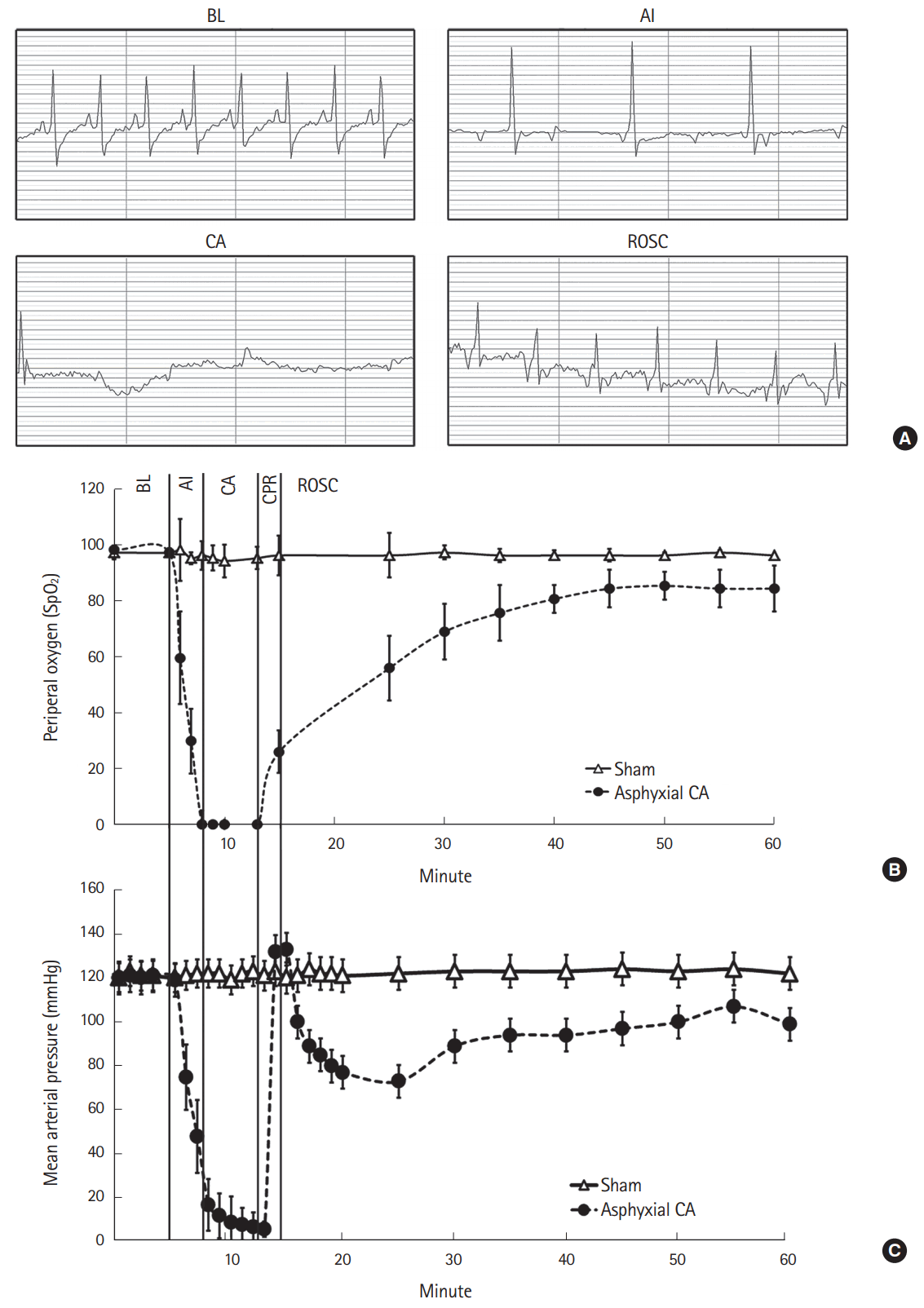
There was no significant difference between baseline characteristics, including body weights, MAP, and SpO2, of rats in the two groups. CA was induced 3 to 4 minutes after intravenous injection of vecuronium bromide (2 mg/kg) and confirmed with isoeletric electrocardiograms and SpO2 and MAP measurements (Fig. 2). Electrocardiograms, SpO2, and MAP changed as expected in the experimental protocol. Baseline and post ROSC physiologic variables are shown in Table 1 (P<0.05 vs. baseline). Body temperatures were the same as those at baseline or after ROSC (Table 1).
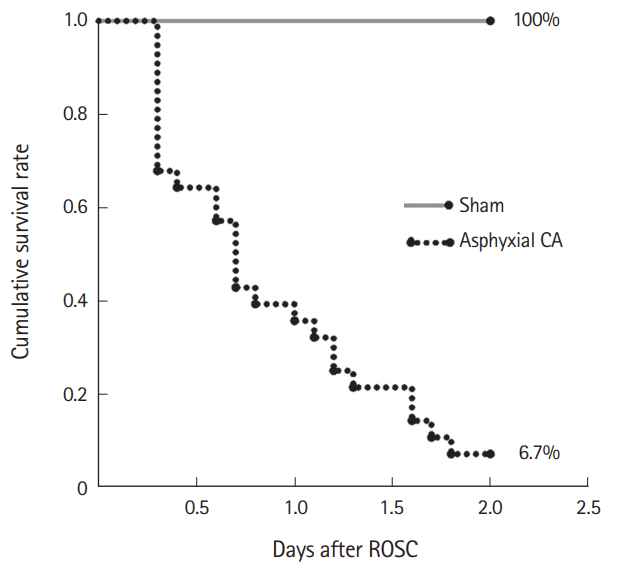
The survival rate in rats was determined 2 days after CA. Results are shown in Fig. 3. Kaplan-Meier analysis showed a severe reduction in survival (P<0.05). The survival rate of rats was 66.7% at 12 hours after ROSC and decreased to 36.7% 1 day after ROSC. Two days after ROSC, the survival rate was 6.7%.
Histopathological changes
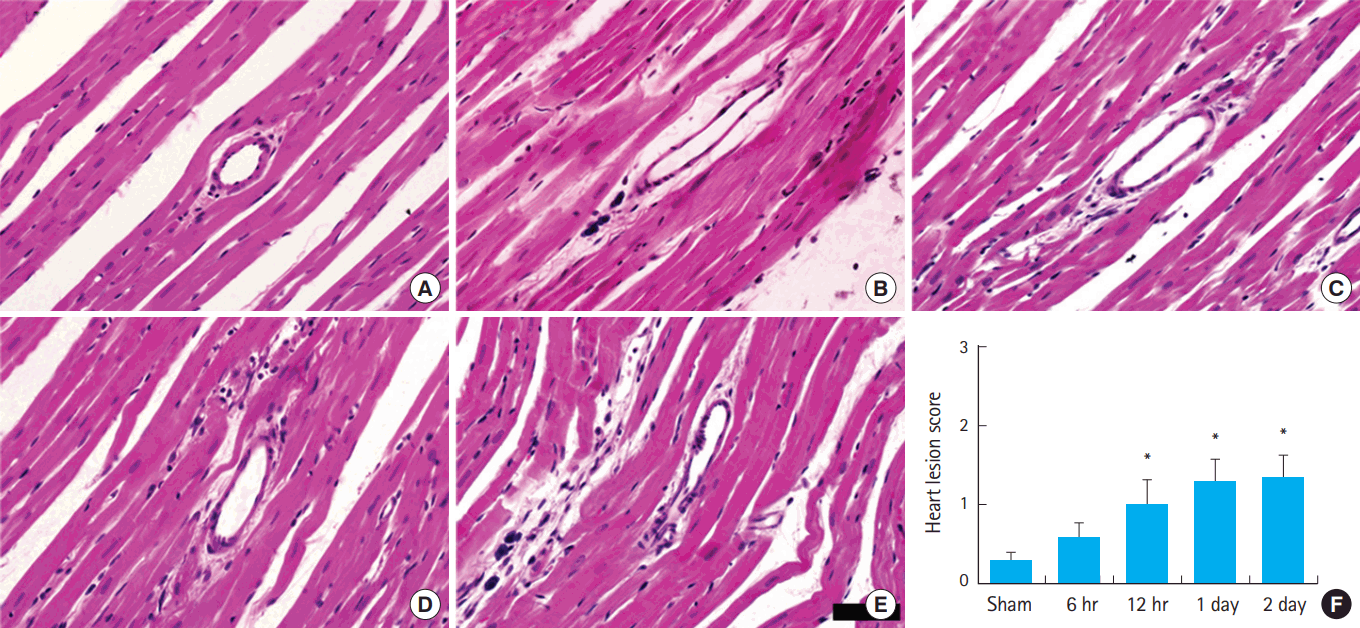
Heart histopathology following CA was examined by H&E staining (Fig. 4). The infiltration of inflammatory cells was apparent in the cardiac parenchyma beginning 6 hours after CA, and degeneration around vessels was found beginning 12 hours after CA. Histopathological scores increased significantly (P<0.05) at 12 hours after CA relative to those in the sham-CA group, and scores were maintained after that time.
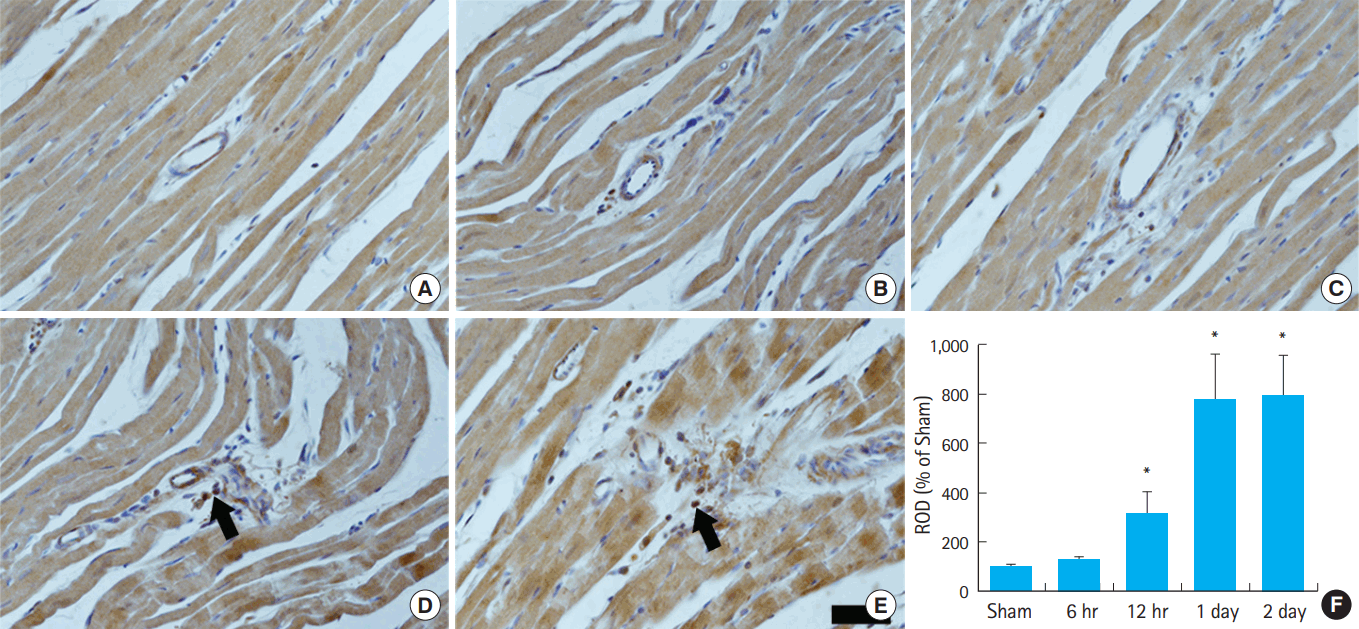
TNF-α immunoreactivity
In this study, the inflammatory response after ROSC was assessed by TNF-α immunohistochemistry (Fig. 5). In the sham-CA group, TNF-α immunoreactivity was low in cardiac tissue, and few TNF-α immunoreactive cells were observed in the connective tissue. However, the number of TNF-α-immunoreactive cells increased significantly (P<0.05) beginning 1 day post CA compared with that in the sham-CA group (Fig. 5).
DISCUSSION
The survival rate of patients with out-of-hospital CAs who were administered CPR by ambulance staff was reported to be 14.6% to 39% in cases who survived through admission, with half of them dying within the first 24 hours in the hospital [15]. Che et al. [12] reported a survival rate of about 40% 2 days after ROSC in a rat model of asphyxial CA, whereas Kida et al. [16] reported that all mice died within 1 day of ROSC in a mouse model of potassium-induced CA. In our present study, survival immediately decreased after ROSC, reaching 6.7% at 2 days after ROSC. As described above, the survival rate following CA in animal models differs according to the methods or animals used. In this study, histopathological evaluations of the heart were performed in the early stage of PCAS because the survival rate was shown to decrease significantly 2 days after ROSC.
In the present study, histopathological scores in hearts following induction of CA increased beginning 12 hours after CA, and scores were maintained after this time. Hayashida et al. [17] reported perivascular and interstitial fibrosis on the endocardial side of the myocardium 1 day after ROSC in a rat model of CA; however, the histological damage did not seem to be severe. On the other hand, in swine studies, coronary blood flow was found not to be reduced in the 30 minutes after ROSC, even though significant dysfunction occurred during this period, indicating a stunning phenomenon rather than permanent injury or infarction [3].
Inflammatory cytokines play important roles in the pathophysiology of PCAS and have been implicated in myocardial and brain dysfunction in the early post-CA period [18,19]. In particular, an increase in pro-inflammatory cytokine levels has been reported following resuscitation [20]. TNF-α level increases shortly after ROSC and is predictive of early death; specifically, plasma levels of TNF-α are inversely correlated with myocardial function after CA [21]. TNF-α, which is a pro-inflammatory cytokine and master regulator of the inflammatory response, is produced primarily by macrophages but also by a broad variety of cells, including lymphoid cells, mast cells, endothelial cells, myocytes, adipose tissue, fibroblasts, and neurons, following CA [18]. Recently, TNF-α protein levels were shown to be increased in the left ventricles of hearts 6 hours after CA [10,21]. In addition, Qi et al. [22] reported a significant increase in TNF-α protein levels in the lungs of an animal model of cardiopulmonary bypass. In our present study, we found that the number of TNF-α immunoreactive cells increased in hearts after CA, although this immunoreactivity and histopathology were not pronounced.
The survival rate of rats with asphyxial CA was very low at 2 days post CA (66.7% at 6 hours, 36.7% at 1 day, and 6.7% 2 days after ROSC following CA). However, histopathological and inflammatory changes in the heart were not pronounced in the early stage after CA. Therefore, the factors that determine the low survival rate after ROSC should be further examined in animal models of CA.