Effect of complement C1-esterase inhibitor on brain edema and inflammation after mild traumatic brain injury in an animal model
Article information

Abstract
Objective
Traumatic brain injury (TBI) is characterized by damage to the blood-brain barrier, inflammation, and edema formation. In this pilot study, we aimed to investigate the effects of a complement inhibitor, C1-esterase inhibitor (C1 INH), on brain edema and inflammation in a rat model of mild TBI.
Methods
Thirty-six male Sprague Dawley rats were randomly assigned to control, TBI, or TBI plus C1 INH groups. TBI and TBI plus C1 INH rats received an injection of saline or 25 IU/kg C1 INH, respectively, with TBI using a weight drop model. Control rats received saline only. Rats were subsequently euthanized and their brain tissue harvested for analysis. The primary outcome was the extent of edema as assessed by the brain’s water content. Secondary outcomes included enzyme-linked immunosorbent assays to determine levels of pro-inflammatory mediators.
Results
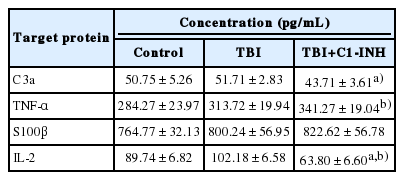
Tumor necrosis factor-α levels were significantly greater in TBI rats than control rats, indicating that inflammation was generated by the weight drop impact. Brain water content following TBI was significantly different between TBI rats treated with C1-INH (78.7%±0.12), untreated TBI rats (79.3%±0.12), and control rats (78.6%±0.15, P=0.001). There was a significant decrease in C3a and interleukin 2 levels among C1 INH–treated rats compared with untreated TBI rats, but no change in levels of tumor necrosis factor-α and S100β.
Conclusion
C1-INH inhibited the complement pathway, suggesting that C1-INH may have a therapeutic benefit in TBI. Further studies are needed to investigate the effect of C1-INH on clinical outcomes.
INTRODUCTION
Traumatic brain injury (TBI) is defined as an alteration in brain function resulting from an external physical force [1]. In the US, a total of 2.8 million TBIs occurred during 2013, with approximately 56,000 deaths [2]. In 2017 and 2018, there was an average of 903 trauma cases per year at our Level 1 trauma institution, of which 246 had a TBI. TBIs pose a significant public health and financial burden, with an estimated 2.5–6.5 million people in the US living with a disability as a consequence of TBI [1,3]. TBI pathophysiology is characterized by primary and secondary brain injury. Primary injury occurs at the time of trauma and leads to tissue damage at the site of injury, disruption of the blood-brain barrier (BBB), axonal injury, and neuronal death [3,4]. These effects initiate a cascade of events with biochemical and metabolic derangements that contribute to secondary tissue damage, including ischemia, neural injury, and hemorrhage [4-6]. Inflammation is an important contributor to secondary injury and damage to the BBB, and along with upregulation of endothelial adhesion molecules, leads to leukocyte infiltration and proinflammatory cytokine release [3,7], as well as edema [8]. The entry of fluid through the damaged endothelium causes an increase in intracranial pressure and subsequent vascular compression, resulting in decreased perfusion and ischemia [9]. Secondary injury is an important determinant of outcomes in patients with TBI [10]. However, unlike primary injury, it is modifiable and has been established as an important target for intervention in mitigating the overall morbidity and mortality of this disease. To date, current methods of management remain limited and are largely supportive.
The complement system, a series of zymogen proteins of the innate immune system, plays an important role in the development of secondary injury in TBI [4]. Upon activation, the enzymes of the complement system can perform a range of immunologic functions, namely opsonization leading to phagocytosis, chemotaxis and stimulation of immune cells, and formation of a membrane attack complex (MAC) which causes cell wall rupture [11]. Immunohistochemical analysis of brain sections from patients with TBI showed elevated levels of activated complement components, specifically C1q, C3b, C3d, and MAC, in the penumbra of the injured area [12]. Similarly, raised levels of complement factors were observed in the cerebral spinal fluid of TBI patients [13,14]. Activation of the complement system occurs early after trauma and is associated with increased mortality rate and organ failure, indicating the importance of the complement system in secondary injury and patient outcomes [15,16].
C1-esterase inhibitor (C1-INH) is a member of the serpin family of protease inhibitors and inactivates a variety of proteases including some involved in complement activation, activation of the contact-kinin system and the fibrinolytic/coagulation system [17]. Previous studies have suggested it may be beneficial in a number of inflammatory disorders, including secondary TBI [18,19]. In this study, we investigated whether C1-INH administration could reduce brain edema and inflammation in TBI in an animal model. To address this, we performed a pilot study using an established weight drop rat model of TBI that results in inflammation, breakdown of the BBB, and edema formation [20,21].
METHODS
Ethics statement
The approval of the Institutional Animal Care and Use Committee (ACC-330) was obtained prior to initiation of the study.
Experimental animals
Thirty-six male, approximately 30-day-old Sprague Dawley rats (Taconic, New York, NY, USA; about 100–200 g weight) were purchased. Rats were of uniform age and sex in order to standardize and minimize variability of the response to C1-INH and TBI. The rats were housed and cared for by the Central Animal Facility staff, who were also responsible for assisting with the preparation, anesthesia, euthanasia, and disposal of the animals.
Study design
Rats were randomly divided into three groups: control (n=12), TBI (n=12), and TBI with C1-INH administration (n=12). The rats were delivered in numbered cages and were sequentially assigned to each group to ensure randomization. At time zero, all rats were individually placed in an induction chamber (EZ-SA800 Single Animal System; E-Z Systems, Palmer, PA, USA), induced with 3% isoflurane for 3 minutes and allowed to reach a deep anesthetic state. Control rats received a 0.9% normal saline solution injection into the tail vein, which was located by warming the tail and aspirating/injecting a total of 0.2-mL normal saline solution with a 27-gauge needle on a 1 mL syringe. TBI rats similarly received a normal saline solution injection following anesthesia and a TBI was induced as described below. TBI with C1-INH rats received an injection of 25 IU/kg of C1-INH (Berinert; CSL Behring GmbH, King of Prussia, PA, USA) following anesthesia, and a TBI was induced (i.e., C1-INH administration and TBI were essentially simultaneous). All rats were allowed to wake undisturbed and were housed and cared for over the course of the following 48 hours. All rats were euthanized with carbon dioxide gas, and their brain tissue was harvested for analysis. One half of each brain was used for the measurement of edema (primary outcome) and the other half was used for the measurement of the secondary outcomes. Two rats were excluded from the final analyses due to experimental error.
TBI apparatus
A TBI apparatus was constructed and adapted from a previously described and validated weight drop model (Fig. 1) [22]. Rats were placed in the prone position on scored tin foil beneath a cylindrical Plexiglass tube, which was positioned 2 cm above the dome of the skull of the rat. A 400-g cylindrical weight was released from one meter and impacted the skull at 90°, generating a force of 3.92 N. As the rats were hovering on the foil placed over soft foam in a collection container, the impact caused a glancing blow intended to cause a mild TBI.

Weight drop model. The anesthetized rat was placed on scored tin foil above a layer of soft foam in a collection container. A plexiglass tube was positioned 2 cm above the rat’s skull and a 400-g weight was released from a height of 1 m.
Outcomes
The primary outcome of the study was the extent of intracerebral edema. Secondary outcomes consisted of the extent of tissue inflammation and injury, including immunohistochemistry to analyze neutrophil infiltration, brain histology to examine tissue injury, and enzyme-linked immunosorbent assays (ELISA) to determine levels of pro-inflammatory mediators and markers of tissue damage.
Determination of brain edema
Brain edema formation was calculated using a wet weight-dry weight method. Half of each brain was weighed shortly after the brain was removed from the skull to determine the wet weight. The samples were then placed in an incubator at 60°C for 72 hours and reweighed to assess the dry weight. Brain water content, i.e., the percent of brain weight attributable to the presence of water, was used as a surrogate for edema and was measured using the following equation: (wet weight [g]-dry weight [g])/wet weight (g).
Histology and immunohistochemistry
A 1-mm coronal section of brain tissue was removed and analyzed by a histopathologist blinded to the treatment group. These sections were fixed with 10% formalin and embedded in paraffin. Coronal slices were taken at various fixed portions of the cortex and stained with hematoxylin and eosin. Immunohistochemistry was also performed with the coronal slices by staining with antineutrophil elastase antibody (AB21595; Abcam, Cambridge, MA, USA), a neutrophil localizing antibody, followed by an anti-rabbit secondary detection antibody. Levels of anti-neutrophil elastase were quantified by determining the number of positive cells per high-power microscopic field.
ELISA analysis
An ELISA was performed to measure levels of tumor necrosis factor (TNF)-α, interleukin (IL)-2, interferon γ, transforming growth factor β1, IL-6, monocyte chemotactic protein 1, intercellular adhesion molecule 1, S100β, and C3a. The proteins were extracted from the rat brain. For each ELISA, commercial kits were used and ELISAs were performed according to the manufacturer’s instructions (Table 1). Briefly, samples were diluted to obtain the same amount of total protein per well and loaded into the wells of a 96-well microplate precoated with the appropriate primary capture antibody (50–100 µg, depending on the kit). Each sample was run in duplicate. A standard of known concentration of the protein was also run in duplicate to enable protein quantification.
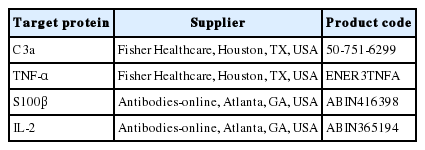
Enzyme-linked immunosorbent assay kits
The microplate was incubated at 37°C for 1 hour to allow the proteins to attach to the microplate. A blocking buffer was added to minimize non-specific binding, after which the microplate was incubated for 1 hour at 37°C. The samples were washed and the primary detection buffer added. The microplate was incubated at room temperature for ≥1 hour. The samples were washed thoroughly with wash buffer, before a secondary antibody, with a colorimetric tag, was added. The microplate was incubated for 30 minutes at 37°C and washed again to remove any unbound antibody. A substrate solution was added and the plate was incubated at room temperature in the dark. The substrate triggered a reaction with the colorimetric tag, causing a color change in the wells containing a bound secondary antibody, thereby indicating the presence of the target protein. A stop solution was added after 10–20 minutes to end the reaction. The microplate was read on a spectrophotometer at 450 nm and 550 nm and the reading at 550 nm was subtracted from the reading at 450 nm. Absorbance measurements were converted to protein concentrations using the equation of the standard curve.
Statistical analysis
Sample size was determined by a power analysis based on previous studies of the effect of C1-INH on edema in animal models of TBI. Results were expressed as means and standard errors of the mean. Data for the primary endpoint were analyzed using an analysis of variance, and secondary endpoints were analyzed using a Student t-test. P-values <0.05 were considered statistically significant.
RESULTS
Intracerebral edema
Measurement of brain water content demonstrated significantly different water content levels between TBI rats (79.3%±0.12), control (78.6%±0.15) and TBI plus C1-INH rats (78.7%±0.12) (P=0.001, between all groups) (Fig. 2).
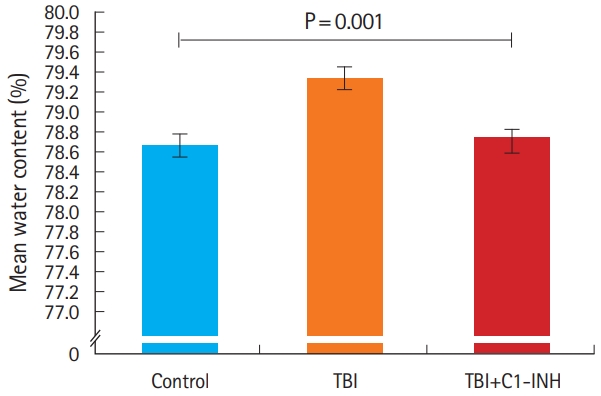
Intracerebral edema, as measured by the percentage of water content averaged across each group and measured from half of each brain. TBI, traumatic brain injury; C1-INH, C1-esterase inhibitor.
Levels of inflammatory mediators by ELISA
Levels of C3a, a product of complement activation, were similar in the TBI and control groups (P=0.44). However, the level of C3a was significantly decreased in TBI plus C1-INH rats compared with the untreated TBI group rats (P=0.048) (Fig. 3 and Table 2).
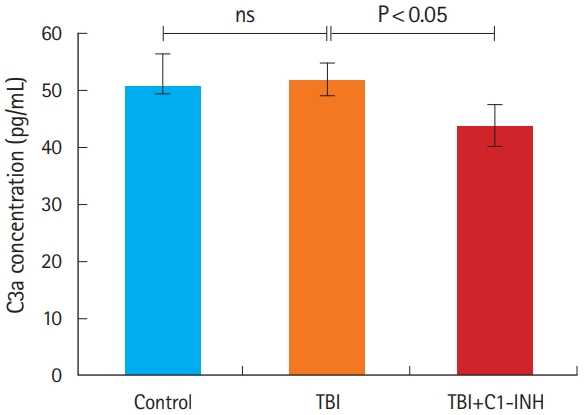
C3a protein concentrations in the brain. ns, not significant; TBI, traumatic brain injury; C1-INH, C1-esterase inhibitor
Concentrations of inflammatory markers as measured by enzyme-linked immunosorbent assays
In all combined TBI rats (TBI and TBI plus C1-INH groups), the mean concentration of TNF-α was significantly higher than in control rats (328.09±12.61 pg/mL vs. 284.27±23.97 pg/mL; P=0.043) (Fig. 4). Similarly, S100β levels were higher in all combined TBI rats (811.43±39.32 pg/mL) compared with control rats (764.77±32.12 pg/mL); however, this difference was not statistically significant (P=0.22) (Fig. 4).

Levels of inflammatory markers in the brain, as measured by enzyme-linked immunosorbent assays. (A) Tumor necrosis factor α and (B)S100β. TBI, traumatic brain injury; ns, non-significant.
There were no significant differences in the concentrations of TNF-α and S100β between TBI and TBI plus C1-INH groups (Table 2). In contrast, the mean concentration of IL-2 was significantly lower in TBI plus C1 INH rats compared with untreated TBI rats (P<0.01), with levels decreasing to below those of the control group (Table 2). ELISAs were also performed to measure levels of IL-6, monocyte chemotactic protein 1, ERIFNG, transforming growth factor β1, C5a, and intercellular adhesion molecule 1, but no significant differences were observed between groups (data not shown).
Immunohistochemistry and histology
Following hematoxylin and eosin staining of coronal sections of the brain near the site of impact, no consistent features of tissue injury or hemorrhage were identified in any rats. Additionally, a neutrophil-localizing stain showed no significant differences among groups in terms of the density of the stain (data not shown).
DISCUSSION
Secondary brain injury is characterized by edema and inflammation, including complement activation, and is known to be an important determinant of outcomes in patients with TBI [3,8,10]. In this study, we found a statistically significant difference in brain water content between control, TBI, and TBI plus C1 INH groups, indicating that the complement inhibitor, C1-INH, may affect edema in rats with a TBI. While the difference was small, per the Monro-Kellie doctrine, even small differences in the rigid, confined intracranial space could have significant clinical consequences on intracranial pressure [23].
This study utilized a weight drop model of TBI in rats; such models are an established technique to study the pathophysiology of TBI by replicating the characteristics of human TBI [20,21]. In the current study, brain water content was quantitatively higher in TBI rats compared with the control group. Furthermore, our study demonstrated an increase in inflammatory markers in the brains of rats experiencing TBI. Indeed, the TNF-α concentration was significantly higher in TBI rats compared with control rats, and levels of S100β were also higher in TBI rats, but not to a significant extent. These results suggest that edema and inflammation were indeed generated by the weight drop impact, supporting the use of this method to model TBI in rats.
This is the first study to investigate the effect of C1-INH on edema, complement activation, and inflammatory mediators simultaneously. Complement has been shown to have an important role in the development of secondary injuries following TBI. Bellander et al. [6] observed activation of the complement system in patients with severe TBI, characterized by an increase in levels of MAC in cerebrospinal fluid. The increase in MAC was paralleled by an increase in the tissue damage markers S100β and neuronspecific enolase, suggesting that complement activation may contribute to additional secondary brain injuries through its membrane-destructive properties, thereby leading to a more pronounced loss of BBB integrity. Similarly, Ganter et al. [15] examined 208 patients with trauma and found that MAC was activated early after trauma. Specifically, the extent of MAC activation was correlated with injury severity, tissue hypoperfusion, and worse clinical outcomes, including increased mortality rate and development of organ failure. Therefore, the complement system represents a potential therapeutic target in TBI, particularly to minimize the development of secondary injuries.
In this study, ELISA analysis showed a significant decrease in C3a levels among C1-INH-treated rats compared with untreated TBI rats, suggesting C1-INH successfully targeted the complement pathway. However, C1-INH did not appear to moderate levels of TNF-α or any of the other inflammatory mediators tested, with the exception of IL-2. Thus, while the decrease in edema following C1-INH administration may in part be related to a reduction in inflammation, this study did not clearly identify which mediators are involved in this process. It should also be noted that neither the treated nor untreated TBI groups showed evidence of neutrophil influx or consistent brain tissue injury, which likely reflects the study design aimed at assessing only mild-tomoderate TBI.
Previous studies have shown that complement inhibitors may be effective in the treatment of TBI in animal models. A C6 antisense RNA, which inhibits MAC deposition, led to reduced inflammation, accumulation of macrophages, and neuronal apoptosis, as well as enhanced neurologic performance versus placebo in TBI mice, supporting the potential of the complement system as a therapeutic target in TBI [24]. C1 INH has been used in a mouse cryolesion model, reducing cortical lesion volumes by nearly 75% when administered 1 hour after trauma onset. C1-INH also stabilized BBB integrity and decreased the infiltration of immune cells and production of pro-inflammatory cytokines in the brain, suggesting that C1 INH may attenuate neurodegeneration and the development of a secondary injury by inhibiting the inflammatory pathway [25]. In addition, in two studies by Longhi et al. [18] and Longhi et al. [19] examining the use of C1-INH in a controlled cortical impact mouse model, C1-INH-treated mice showed attenuated neurologic motor deficits and cognitive dysfunction compared with control mice receiving saline, with a significantly improved ability to learn after 4 weeks. The mice also showed reduced brain tissue damage compared with control mice. However, while these effects were observed when C1-INH was administered 10 minutes post-injury, mice receiving C1-INH at 1 hour post-injury showed no significant improvement, suggesting that the timing of treatment is critical [19]. These findings support the study of C1-INH as a possible treatment for patients with TBI, suggesting that the inhibitor may limit secondary injury by reducing inflammation, thereby improving cognitive and neurologic outcomes. These studies also suggest that the timing of C1-INH administration used in our study (prior to weight drop impact) may have had an impact on the efficacy of the drug used to reduce inflammation. Repeating the current study with C1-INH administered at different time points following TBI may improve our understanding of the effects of the timing of its administration.
While the current study investigated edema and inflammation of the brain following TBI, other studies have assessed the molecular consequences of C1-INH administration in animals with extra-neurologic inflammation. Two studies in swine found that C1-INH led to reduced cardiac, lung, renal, and intestinal tissue damage and improved survival vs control animals. C1-INH was associated with lower levels of pro-inflammatory cytokines and reduced complement activation and tissue deposition [26,27], suggesting that C1-INH may be an effective treatment for secondary TBI.
Although these studies suggest that the benefits of C1-INH may be due to inhibition of the complement system, another study suggests that C1-INH may act primarily by inhibiting the kinin and coagulation cascades [28]. These authors found that rats pretreated with C1-INH before ischemia and reperfusion had significantly less edema in the muscle alongside improved muscle viability, with no significant difference in the deposition of complement components [28]. While this study conflicts with our finding that complement activation may be inhibited by C1-INH (as shown by reduced C3a levels in treated vs untreated TBI rats), it highlights the need to better understand the mechanisms involved in secondary brain injury, which will help the development of new therapies.
The use of water content (specifically percent water content, calculated as [wet weight-dry weight]/wet weight) as a surrogate marker for brain edema is well established in animal models. However, it should be noted that there are issues with this approach. The relationship between percent water content and either water content (g/g dry weight) or brain swelling is not linear, and relatively small changes in percent water content may be associated with notably larger changes in tissue water/swelling [29]. Therefore, the true extent of tissue swelling/brain edema in the current study remains unclear.
Despite the use of animal models in many studies to investigate C1-INH treatment in TBI, they are subject to several limitations. Importantly, there is a lack of homology between rat and human complement, which may affect the efficacy of the drug and result in a different response between rats and humans. For example, the amino acid sequence of rat C5a, levels of which were not affected by C1-INH treatment in our study, is substantially different to the human form (35% non-identical sequence). Such sequence differences are thought to contribute to the enhanced potency of rat C5a compared with C5a of other species [30]. In addition, while the weight drop model is intended to cause TBI and has been shown to mimic many of the characteristics of clinical TBI [20,21], it may not accurately recreate the forces involved in different types of brain injuries, such as acceleration/deceleration, blast, and penetrating trauma. While our study has shown C1 INH to have a positive effect on brain edema levels following trauma, it is unknown how this would translate to cognitive outcome and survival. Indeed, this was a pilot study with limited resources and funding; additionally, neither neuronal injury nor behavioral functional changes were assessed here. Future studies are intended to assess the efficacy of treating TBI with C1-INH at varying doses at different intervals to evaluate the effects of dosing and timing of drug administration in relation to injury on edema and inflammation, as well as the treatment effects of C1-INH on TBI of varying degrees of severity. As part of those studies, it is necessary to measure additional inflammatory mediators to further explore the effect of complement inhibition on the inflammatory response in the brain, in addition to measuring other indicators of inflammation (i.e., apoptosis signaling, and plasmin and kallikrein activation/inhibition). In addition, future studies are needed to investigate the clinical response to C1-INH treatment and improve understanding of the role of complement in secondary brain injury.
In summary, C1-INH treatment in a rat model of TBI leads to reduced complement activation and potentially affects levels of brain edema, suggesting that C1-INH may have a potential therapeutic benefit in TBI. However, with the exception of IL-2, these changes were not associated with reduced levels of inflammatory mediators, suggesting that C1-INH, as administered in this experiment, was insufficient to inhibit the inflammatory response. Although previous studies have demonstrated the benefits of C1-INH in minimizing secondary injuries in TBI animal models, further studies are needed to examine its effect on both edema and inflammation, as well as its consequences on clinical outcomes.
Notes
Laurel Omert is an employee of CSL Behring. No other authors have conflicts of interest to report.
Acknowledgements
Project funding was obtained through Albert Einstein Society $10k Seed Grant no. 8579.
Medical writing support in the form of developing drafts based on author input and editorial assistance was provided by Leigh O’Connor of Fishawack Communications Ltd., and was funded by CSL Behring.
References
Article information Continued
Notes
Capsule Summary
What is already known
Traumatic brain injury (TBI) poses a significant public health and financial burden. Primary TBI occurs as a result of the initial impact. Secondary TBI follows the occurrence of primary TBI, and is characterized by edema and inflammation, including complement activation. Secondary TBI is an important determinant of clinical outcomes and is modifiable. However, current treatment options are limited.
What is new in the current study
This is the first study to investigate the effect of C1-esterase inhibitor (C1-INH) on edema, complement activation, and inflammatory mediators simultaneously. C1-INH led to inhibition of the complement pathway in a rat model of TBI, highlighting the potential therapeutic benefits of C1-INH in secondary TBI.