INTRODUCTION
Modern cardiopulmonary resuscitation (CPR) was introduced more than 50 years ago [1]. Several pharmaceutical and non-pharmaceutical modalities have been investigated to improve the outcomes. However, most of these modalities have not shown any clinical benefits. Nonetheless, novel therapeutic modalities should be further explored to discover a treatment that would benefit patients that experience cardiac arrest, who continue to have poor outcomes in the current era [2].
Pralidoxime (2-pyridine aldoxime methyl chloride) belongs to a family of compounds called oximes. As pralidoxime can reactivate the organophosphate-inhibited cholinesterase enzyme, it has been widely used to treat organophosphate poisoning. However, minimal attention has been given to the other pharmacological properties of this compound. In the late 20th century, several studies reported that pralidoxime had a stimulatory action on vascular smooth muscle in addition to its ability to reactivate the inhibited cholinesterase enzyme [3-7]. Its ability to constrict blood vessels and hence elevate arterial pressure may aid in successful resuscitation by increasing the coronary perfusion pressure (CPP), which is a major determinant of myocardial blood flow during CPR and of restoration of spontaneous circulation (ROSC) [8,9]. Pralidoxime may also facilitate successful resuscitation by reducing the ischemic contracture, thus improving the hemodynamic efficacy of CPR. 2,3-Butanedione monoxime (BDM) has been found to reverse ischemic contracture and improve resuscitability in a pig model of cardiac arrest [10,11]. Both BDM and pralidoxime belong to the oxime family and have common mechanisms of action [12]. Thus, pralidoxime may also reverse ischemic contracture in a manner similar to that of BDM. However, to our knowledge, no study on the effects of pralidoxime administration during CPR has been conducted.
In this study, we investigated the effects of pralidoxime administration during CPR in a pig model of out-of-hospital cardiac arrest with a focus on ischemic contracture and CPP. We hypothesized that pralidoxime would reduce the ischemic contracture and improve the CPP.
METHODS
This prospective randomized study involved 16 Yorkshire/Landrace cross pigs weighing 25.2±2.9 kg. The Animal Care and Use Committee of Chonnam National University approved the protocol for this study (CNU IACUC-H-2017-18). Animal care and experiments were in accord with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Animal preparation
After the intramuscular administration of pre-medications (ketamine [20 mg/kg] and xylazine [2.2 mg/kg]), the animals were placed in a supine position in a U-shaped trough and orally intubated. Anesthesia was provided using 70%:30% N2O:O2 and 0.5% to 2% sevoflurane, which was titrated to prevent signs of pain (reactive and wide pupils, tachycardia, and hypertension). Ventilation rates were adjusted to achieve normocapnia. A double-lumen catheter was inserted via the left femoral artery for blood pressure monitoring and blood sampling. The right external jugular vein was cannulated using a 7-F introducer sheath to monitor right atrial (RA) pressure and to insert a right ventricle pacing wire. An end-tidal carbon dioxide (ETCO2) sample line (B40 Patient Monitor; GE Healthcare, Chalfont St. Giles, UK) was attached to the ventilator circuit. To obtain an adequate left ventricular (LV) long-axis view during CPR, a skin incision was made to the right of the xiphoid process, and a pocket extending 4 to 5 cm under the right side of the sternum was made wide enough to ensure the free passage of a transesophageal echocardiography (TEE) probe (UST-5293-5; Hitachi Aloka Medical, Tokyo, Japan) [10,11]. Then, the TEE probe was precordially inserted via the pocket, and the best obtainable long-axis view of the LV was sought using the manipulative controls of the TEE probe (Fig. 1). In our previous experiment, in which the TEE probe was advanced into the esophagus, chest compression precluded the echocardiographic visualization of the LV. However, with this method, no interruption in chest compression was required to visualize the LV during CPR. During the preparation phase, saline was administered to maintain an RA pressure of 8 to 12 mmHg. After the baseline measurements were obtained, vecuronium (0.05 mg/kg) was administered intravenously to inhibit the potential confounding effect of gasping.
Experimental protocol
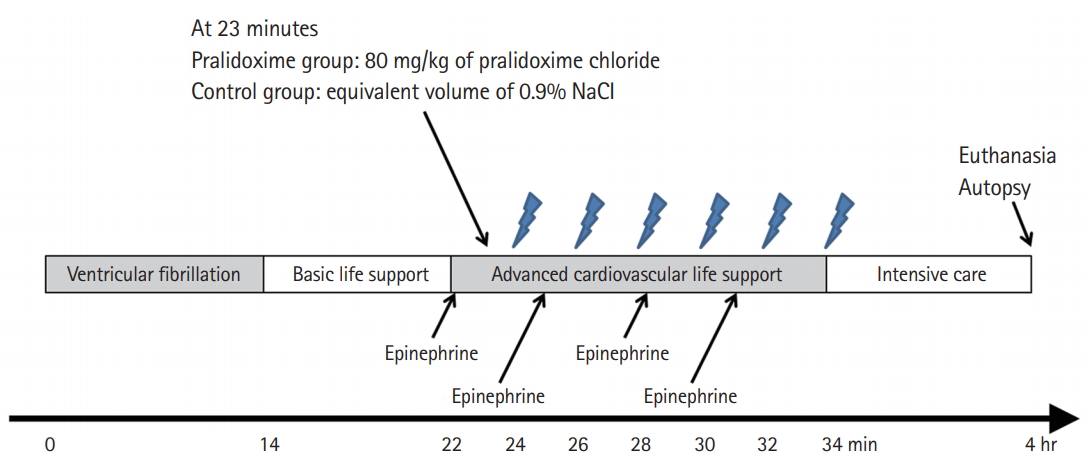
The experimental timeline is shown in Fig. 2. Ventricular fibrillation (VF) was induced by applying an electrical current (60 Hz and 30 mA of an alternating current) via a right ventricle pacing wire. Immediately after inducing VF, an investigator who was otherwise uninvolved with this study opened a sealed envelope that assigned animals to either the control group or the pralidoxime group; the saline placebo and the pralidoxime solution (80 mg/kg of pralidoxime chloride; JW Pharmaceutical, Seoul, Korea) were prepared in equal volumes (20 mL). In a previous study on the cardiovascular effects of various doses of pralidoxime (5–40 mg/kg) in open-chest dogs [5], the absolute maximum response was not determined. Thus, a dose of 80 mg/kg was used, which is twice the highest dose used in the previous study. All other investigators remained blinded to the treatment allocation.
After 14 minutes of untreated VF, basic life support (BLS) with cycles of 30 chest compressions followed by two ventilations with ambient air was started. After 8 minutes of BLS, advanced cardiovascular life support (ACLS) based on the recent resuscitation guidelines was started [13]. Chest compressions were delivered at a rate of 100/min using a piston-driven chest compression device (Life-Stat; Michigan Instruments, Grand Rapids, MI, USA). During ACLS, the compression depth was adjusted to decrease the anterior–posterior diameter of the chest by 20%. In our previous study [14], this depth was the maximum depth for avoiding critical injuries related to CPR. Meanwhile, 70% of this depth was used for compressions during BLS to simulate the suboptimal quality of chest compressions in out-of-hospital CPR [15,16]. During ACLS, positive-pressure ventilations with high-flow oxygen (14 l/min) were provided at a rate of 8/min using a volume-marked bag devised by Cho et al. [17] One minute after ACLS was started, either pralidoxime (pralidoxime group) or saline placebo (control group) was administered into the RA. During ACLS, 0.02 mg/kg of epinephrine was administered every 3 minutes, and defibrillation was attempted using a single biphasic 150-J electric shock at 2-minute intervals if indicated. If ROSC was not achieved within 12 minutes of ACLS, resuscitation efforts were discontinued.
Animal that achieved ROSC were mechanically ventilated and then underwent a 4-hour period of intensive care. Throughout the intensive care period, titrated doses of sevoflurane were administered to maintain adequate anesthesia. At the end of the 4-hour period, the animal was euthanized using an infusion of potassium chloride.
Measurements
The primary outcomes were CPP and LV ischemic contracture, which was assessed by measuring the LV wall thickness and LV chamber area. Aortic pressure and RA pressure were continuously monitored (CS/3 CCM; Datex-Ohmeda, Helsinki, Finland), and the data were transferred to a personal computer using S/5 Collect software (Datex-Ohmeda). Systolic aortic pressure was defined as the peak aortic pressure during the chest compression phase. Diastolic aortic pressure was defined as the lowest inflection point at the beginning of the next compression-induced pressure upstroke. CPP was calculated by subtracting the RA end-diastolic pressure from the simultaneous aortic end-diastolic pressure. Aortic pressure, RA pressure, and CPP were sampled by averaging pressures from five consecutive compressions at 2-minute intervals during BLS and at 1-minute intervals during ACLS. ETCO2 values were determined every 1 minute by averaging the ETCO2 values for the preceding 30-second interval. Echocardiography was performed by a researcher blinded to the treatment allocation at the pre-arrest baseline (5 minutes before the induction of VF), during untreated VF (1, 7, and 14 minutes after the initiation of VF), and during CPR (every 2 minutes during BLS and every 1 minute during ACLS). The LV chamber area and LV wall thickness during CPR were measured using a technically satisfactory LV long-axis view at the frame showing the maximum chamber dimensions of the LV following release of chest compression. The LV chamber area was measured by electronic integration after manual tracing of the endocardial borders. The LV wall thickness was measured in the lateral wall at the mid-ventricular level.
Statistical analysis
The sample size was calculated to detect a difference in the LV chamber area between the groups based on data from a previous study [11]. We calculated a sample size of seven animals in each group to reach an α of 0.05 and a power of 80%. To minimize any effect of data loss, eight animals were included in each group. Continuous variables were tested for normality using the Shapiro-Wilk test. Normally distributed variables were expressed as means±standard deviations, and the independent t-test was performed. Variables that were not normally distributed were expressed as medians with interquartile ranges, and the Mann-Whitney U-test was conducted. Repeated-measures analysis of variance (ANOVA) was used for the comparison of echocardiographic and hemodynamic variables during the untreated VF and BLS. These variables were not fully obtained in one animal because the animal achieved ROSC after 8 minutes of ACLS. Mixed-model analysis, which can retain cases with missing data points, was thus used to compare these variables during ACLS. If a significant group effect was observed, post-hoc comparisons between the groups at each time point were performed using the pairwise t-tests. Data were analyzed using PASW Statistics ver. 18.0 (SPSS Inc., Chicago, IL, USA). A P-value of <0.05 was considered significant.
RESULTS
No significant differences between groups were observed in the baseline measurements (Table 1). Fig. 3 shows the LV wall thickness and LV chamber area. The repeated-measures ANOVA of the LV wall thickness and the LV chamber area during the untreated VF and BLS revealed no significant group effects, time effects, or group-time interactions. During ACLS, the LV wall thickness progressively increased and the LV chamber area progressively decreased. Mixed-model analyses of the LV wall thickness and LV chamber area during ACLS revealed significant time effects (P<0.001 for both the LV wall thickness and the LV chamber area). However, neither group effects nor group-time interactions were observed.
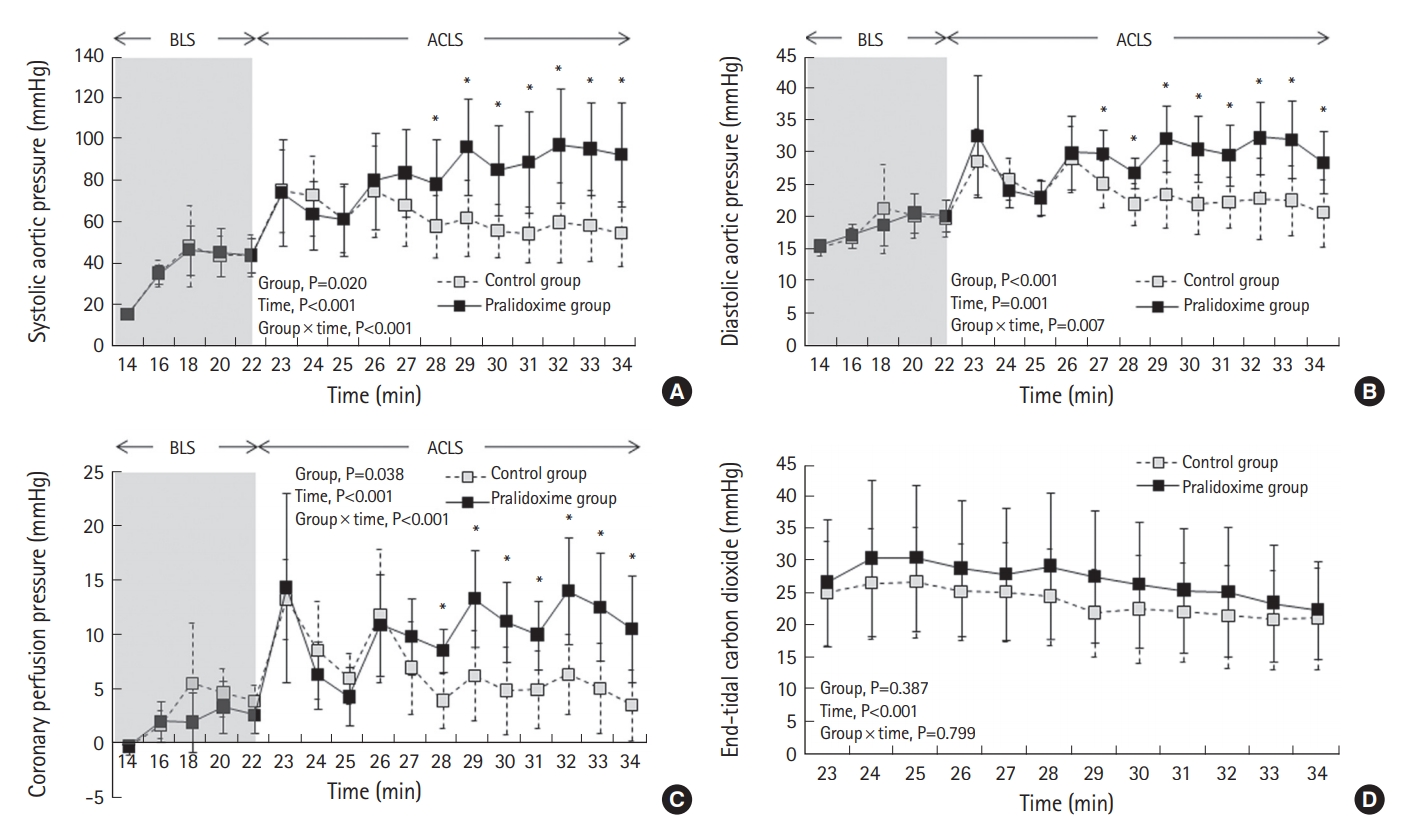
Fig. 4 depicts the hemodynamic variables during CPR. The repeated-measures ANOVA of the systolic aortic pressure, diastolic aortic pressure, and CPP during BLS revealed no significant group effects or group-time interactions, but it revealed significant effects with respect to time (P<0.001 for both the systolic aortic pressure and the diastolic aortic pressure and P=0.001 for CPP). Mixed-model analyses of the systolic aortic pressure, diastolic aortic pressure, and CPP during ACLS revealed significant group effects (P=0.020 for the systolic aortic pressure, P<0.001 for the diastolic aortic pressure, and P=0.038 for CPP), time effects (P<0.001 for both the systolic aortic pressure and CPP and P=0.001 for the diastolic aortic pressure), and group-time interactions (P<0.001 for both the systolic aortic pressure and CPP and P=0.007 for the diastolic aortic pressure). The post-hoc analyses revealed significant differences in the systolic aortic pressure and CPP between the two groups after 28 minutes. The diastolic aortic pressure differed between the groups after 27 minutes. The mixed-model analysis of ETCO2 during ACLS revealed a significant time effect (P<0.001). However, no group effect or group-time interaction was observed with respect to ETCO2.
No animal, except one in the pralidoxime group, achieved ROSC. Thus, in the present study, the rate of ROSC did not differ between the two groups. The successfully resuscitated animal was hemodynamically stabilized without inotropic or vasopressor support and survived the 4-hour intensive care period.
DISCUSSION
To our knowledge, this is the first study to report the effects of pralidoxime administration in an in vivo cardiac arrest model. In a pig model of cardiac arrest, pralidoxime administered during CPR did not reduce the ischemic contracture. However, its administration significantly improved the aortic pressure and CPP.
Although pralidoxime has never been used as a vasopressor in clinical practice, its pressor effect has long been reported in previous studies [3-7]. In 1964, Calesnick et al. [18] reported a substantial elevation in systolic and diastolic blood pressure immediately after intravenous administration of pralidoxime in healthy human participants. However, the mechanism of its pressor action remains controversial. Several researchers have suggested that the vasopressor effect of pralidoxime is mediated through the stimulation of α-adrenergic receptors in the peripheral vessels [3-5]. In a study by Stavinoha et al. [4] that investigated the cardiovascular effects of pralidoxime in anesthetized dogs, pralidoxime produced a significant increase in the systemic arterial pressure, whereas α-adrenergic blockers, such as phentolamine, effectively blocked the pressure response to pralidoxime. Meanwhile, Carrier et al. [6] reported that phentolamine did not affect the response of the aorta to pralidoxime in an isolated aortic strip preparation. They suggested that the cardiovascular effect of pralidoxime was related to an alteration in intracellular calcium storage rather than the α-adrenergic receptor stimulation.
Current resuscitation guidelines recommend the intravenous administration of epinephrine every 3 to 5 minutes during ACLS [13]; thus, repeated epinephrine administration is required in cases of prolonged CPR. Several previous studies have reported that, in contrast to the first epinephrine dose, subsequent epinephrine doses fail to increase CPP above a threshold that is needed for successful resuscitation [19-21]; however, the exact mechanism of this effect is still not proven. Consistent with these studies, the pressor effect of epinephrine in the control group was markedly attenuated with repeated doses in the current study. In the control group, CPP sharply increased following the first and second doses of epinephrine, whereas no sharp increases in CPP were observed after the next epinephrine doses. In the present study, pralidoxime administration abolished the waning of the epinephrine effect, and as a result, CPP after the third and fourth epinephrine doses in the pralidoxime group was significantly higher than that in the control group. Stavinoha et al. [4] reported that pralidoxime potentiated the duration and intensity of the pressor response elicited by epinephrine in anesthetized dogs, which is consistent with the observations in the present study.
Ischemic contracture, which results from cross-bridge formation between actin and myosin following severe adenosine triphosphate depletion [22,23], frequently occurs during prolonged cardiac arrest and compromises the hemodynamic effectiveness of CPR by reducing the LV chamber volume [24,25]. We previously have reported that BDM reduced the ischemic contracture and improved the resuscitability in a pig model of cardiac arrest [10,11]. Because both pralidoxime and BDM are cholinesterase reactivators and share several common mechanisms of action [12], we initially expected that both drugs would have similar effects on the ischemic contracture. However, unlike BDM, pralidoxime had no effect on ischemic contracture in the present study. Atoda et al. have reported similar findings [7]. They have reported that BDM decreased the contractile force and the action potential duration, whereas pralidoxime tended to increase these parameters in the ventricular muscles of guinea pigs [7]. The reasons these two oximes act differently are not clear. However, we postulate that one of the reasons could be related to the difference in their effects on intracellular ionized calcium concentration, which plays an important role in myocardial contraction. BDM reduces the release of calcium from the sarcoplasmic reticulum [26,27], whereas pralidoxime decreases the rate of calcium uptake by the sarcoplasmic reticulum and thus increases the level of ionized calcium available for contractile proteins [6].
Although pralidoxime did not improve the resuscitation rate in the present study, we believe that pralidoxime administration during CPR should be further evaluated. In the present study, CPP did not reach the level that renders successful resuscitation likely even in the pralidoxime group [28,29]. As a result, most of the animals in the present study could not be resuscitated. We chose a prolonged period of untreated VF followed by a prolonged duration of BLS to achieve the pronounced ischemic contracture needed to monitor the treatment effects. Thus, the duration of ischemia until the administration of epinephrine was markedly prolonged, and this might have substantially weakened the effect of epinephrine. Several researchers have suggested that the pressor effect of epinephrine is attenuated when its administration is late during resuscitation because acidosis limits the ability of epinephrine to induce vasoconstriction [20,30]. Given that a higher CPP is associated with successful resuscitation [9,28,29], pralidoxime’s ability to improve CPP may aid in successful resuscitation in cardiac arrest models with a relatively brief duration of untreated VF, during which the epinephrine effect is less attenuated.
Our study has several limitations. First, unlike the majority of patients who experience cardiac arrest, our pigs were young, healthy, and free of atherosclerotic disease. Their response may differ from humans with atherosclerosis. Second, we used a swine model with a significantly prolonged duration of VF before resuscitation. Studies using a different model with a shorter period of untreated VF might obtain different results. Third, the effects on long-term survival and neurologic outcome could not be examined. Fourth, this study lacks the dose-response data. Thus, we are not able to report the optimal dose of pralidoxime. Fifth, no adjustment for multiple comparisons was performed since the analyses were exploratory in nature. Considering the above limitations, the findings of this study should be further investigated.
In conclusion, pralidoxime administered during CPR did not reduce ischemic contracture in a pig model of prolonged out-of-hospital cardiac arrest. However, its administration significantly improved the aortic pressure and CPP. Hence, the present study suggests that pralidoxime potentiates the vasopressor effect of epinephrine administered during CPR. Considering the markedly prolonged ischemic duration used in this study, further studies are required to determine the effect of pralidoxime on resuscitation outcomes.