INTRODUCTION
Ischemia-reperfusion (IR) injury refers to a series of clinical and experimental consequences triggered by tissue ischemia from insufficient oxygen supply, followed by subsequent reperfusion [1,2]. IR injury is implicated in various clinical diseases, including myocardial infarction, stroke, and limb ischemia [3ŌĆō5]. The process of reperfusion following ischemia stimulates the formation of reactive oxygen species (ROS) and oxidative stress, resulting in various inflammatory responses, leading to local tissue injury and remote organ dysfunction [6,7].
Kallistatin is an endogenous serine proteinase inhibitor that binds to the kallikrein protein and inhibits kallikrein activity [8,9]. Kallistatin exhibits various functions such as antioxidation, anti-inflammation, and antiangiogenesis activities [10]. The antioxidative property of kallistatin is accomplished by attenuating ROS formation through inhibition of the oxidase activity of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) [11]. Furthermore, kallistatin promotes the synthesis of endothelial nitric oxide synthase (eNOS), sirtuin 1, and forkhead box protein O1 enzymes, thereby reducing oxidative stress [11ŌĆō14]. As a result of its antioxidative property, kallistatin has a protective effect against IR injury in murine myocardial and renal IR injury models [15,16]. In our previous study [17], the association between low-serum kallistatin level and poor neurological outcomes in out-of-hospital cardiac arrest patients was revealed through a proteomics approach. The study suggested that kallistatin deficiency could potentially diminish the endogenous antioxidative defense capacity of nerve cells, leading to increased neuronal damage and unfavorable neurological outcomes following cardiac arrest.
We hypothesized that kallistatin deficiency would enhance cell death caused by IR injury, possibly resulting from the attenuation of the eNOS-promoting effect of kallistatin. To validate this hypothesis, we conducted experiments using human umbilical vein endothelial cells (HUVECs) in which kallistatin expression was suppressed by ribonucleic acid interference. An in vitro model mimicking IR injury was created by applying oxygen-glucose deprivation and reoxygenation (OGD/R) to HUVECs. The primary objective of this study was to investigate the antioxidant mechanism of kallistatin in the prevention of IR injury.
METHODS
Cell culture
HUVECs (Cat. C2517AS, Lonza) were cultured in EGM-2 Endothelial Cell Growth Medium-2 BulletKit (Cat. CC3162; Lonza) containing 10% fetal bovine serum. The HUVECs were incubated for 24 hours at 37 ┬░C in a humidified atmosphere with 95% air and 5% carbon dioxide. After reaching 90% confluence, the cells were seeded on 96-well plates (2├Ś104 cells/well) or 6-well plates (2├Ś106 cells/well) for subsequent experiments.
Small interfering ribonucleic acid transfection
To generate SERPINA4 knockdown cells, HUVECs were transfected with small interfering RNA (siRNA) targeting the human kallistatin gene SERPNA4. The ON-TARGETplus human SERPINA4 (5267) siRNA-SMARTpool (25 nM, target sequence: 5ŌĆÖ-GGUGAGAGUUGCAGUAACA-3ŌĆÖ, 5ŌĆÖ-CAAUCC-AGCUUAUCAACGA-3ŌĆÖ, 5ŌĆÖ-CCACCAGCUUCGCGAUCAA-3ŌĆÖ, 5ŌĆÖ-GCAAAAUG-AGGGAGAUUGA-3ŌĆÖ; Cat. L016371000050, Dharmacon) and siGLO RISC-Free Control siRNA (Cat. D0016000105, Dharmacon) as control were used for transfection. HUVECs were treated with these siRNAs using DharmaFect1 transfection reagent (Cat. T2001-03, Dharmacon) aaccording to the manufacturer's instructions for 6 hours and then cultured for 18 hours in complete media before further experiments.
Oxygen-glucose deprivation and reoxygenation
Transfected HUVECs were exposed to 90 minutes of OGD followed by 22.5 or 46.5 hours of reoxygenation. To induce OGD, complete medium was replaced with Dulbecco's Modified Eagle Medium without glucose (Cat. 11966025, Thermo Fisher Scientific), and the cells were placed in a hypoxic chamber (Cat. INCO108, Memmert) with a gas mixture of 95% nitrogen and 5% carbon dioxide. After 90 minutes of OGD, the medium was changed back to the complete medium, and the cells were returned to the chamber with 95% air and 5% carbon dioxide for 22.5 or 46.5 hours of reoxygenation.
Cell viability assay
To determine the optimal duration of OGD/R with significant effects on cell survival, cell viability was assessed 22.5 or 46.5 hours after reoxygenation using Cell Counting Kit-8 (CCK-8; Dojindo Laboratories), which uses the tetrazole assay method with modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. After 24 or 48 hours of OGD/R, CCK-8 solution (10 ╬╝L/well) was added and the cells were incubated for 2 hours at 37 ┬░C. Subsequently, the absorbance at a wavelength of 450 nm was measured. We also assessed the cell viability of HUVECs treated with control or SERPINA4 siRNA and exposed to OGD/R using the methods described above to evaluate the effect of SERPINA4 knockdown on OGD/R.
Measurement of kallistatin, eNOS, and total NO concentration
To evaluate the effect of SERPINA4 knockdown on kallistatin expression and OGD/R, the kallistatin concentration in the culture medium was measured with the SERPINA4 Human ELISA Kit (Cat. KA3892, Abnova). To investigate the association between the eNOS-promoting property of kallistatin and its protective effect against IR injury, we measured the eNOS and total NO concentrations. The eNOS concentration in the culture medium was measured using the Human eNOS ELISA Kit (Cat. MBS8291345, MyBioSource), while the total NO concentration in the culture medium was measured using the Total Nitric Oxide and Nitrate/Nitrite Parameter Assay Kit (Cat. KGE001, R&D Systems).
Statistical analysis
To compare cell viability and concentrations of kallistatin, NO, and eNOS among experimental groups, we conducted one-way analysis of variance with Tukey post hoc test. Data were reported as mean┬▒standard error. Statistical analysis was performed using IBM SPSS ver. 27.0 (IBM Corp). The level of statistical significance was set at P<0.05.
RESULTS
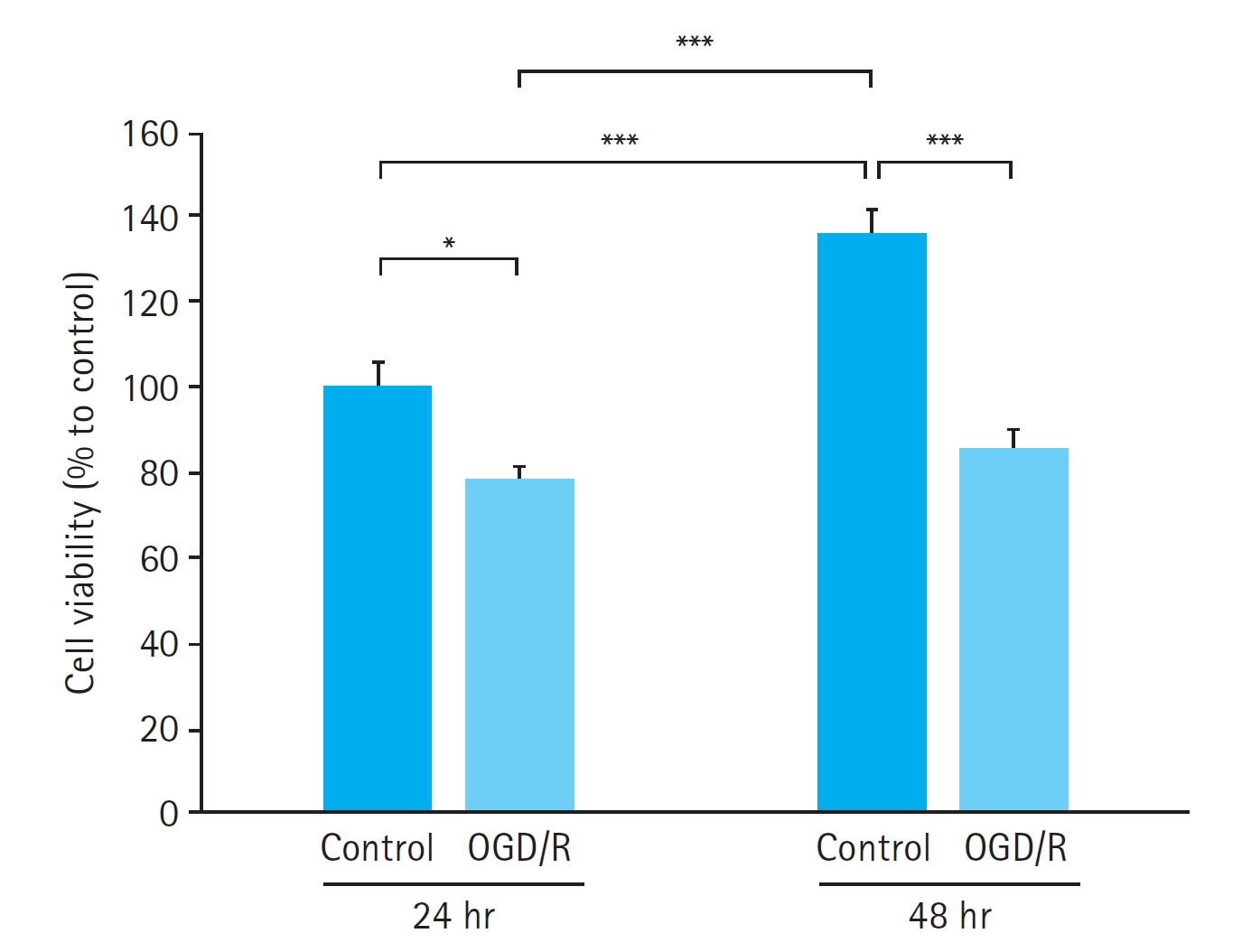
Time-dependent effect of OGD/R on HUVECs
HUVECs exposed to 90 minutes of OGD followed by either 22.5 or 46.5 hours of reoxygenation exhibited reduced viability compared with control cells. The decrease in cell viability after OGD/R was significant at both 24 hours (P<0.05) and 48 hours (P < 0.001), with a more pronounced effect at 48 hours (Fig. 1). As 24 hours of OGD/R was sufficient to induce a significant decrease in cell viability, we selected 24 hours of exposure for further experiments.
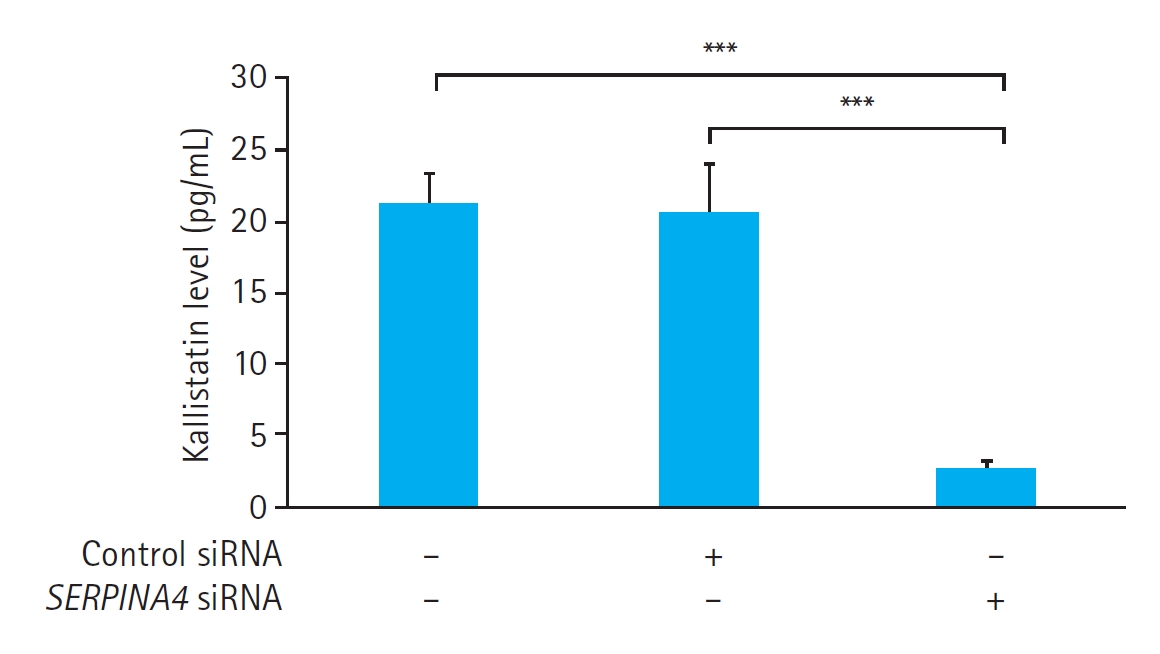
Effects of SERPINA4 siRNA transfection on kallistatin expression in HUVECs
The kallistatin concentration in the culture medium did not show any significant differences between the control cells without any treatment (21.1┬▒2.1 pg/mL) and the cells transfected with control siRNA (20.5┬▒3.3 pg/mL, P=0.981). However, HUVECs transfected with SERPINA4 siRNA demonstrated a significantly lower kallistatin concentration in the culture medium (2.5┬▒0.7 pg/mL) compared with both the control cells (P<0.001) and the cells transfected with control siRNA (P<0.001) (Fig. 2). These results confirmed the successful SERPINA4 knockdown by SERPINA4 siRNA transfection.
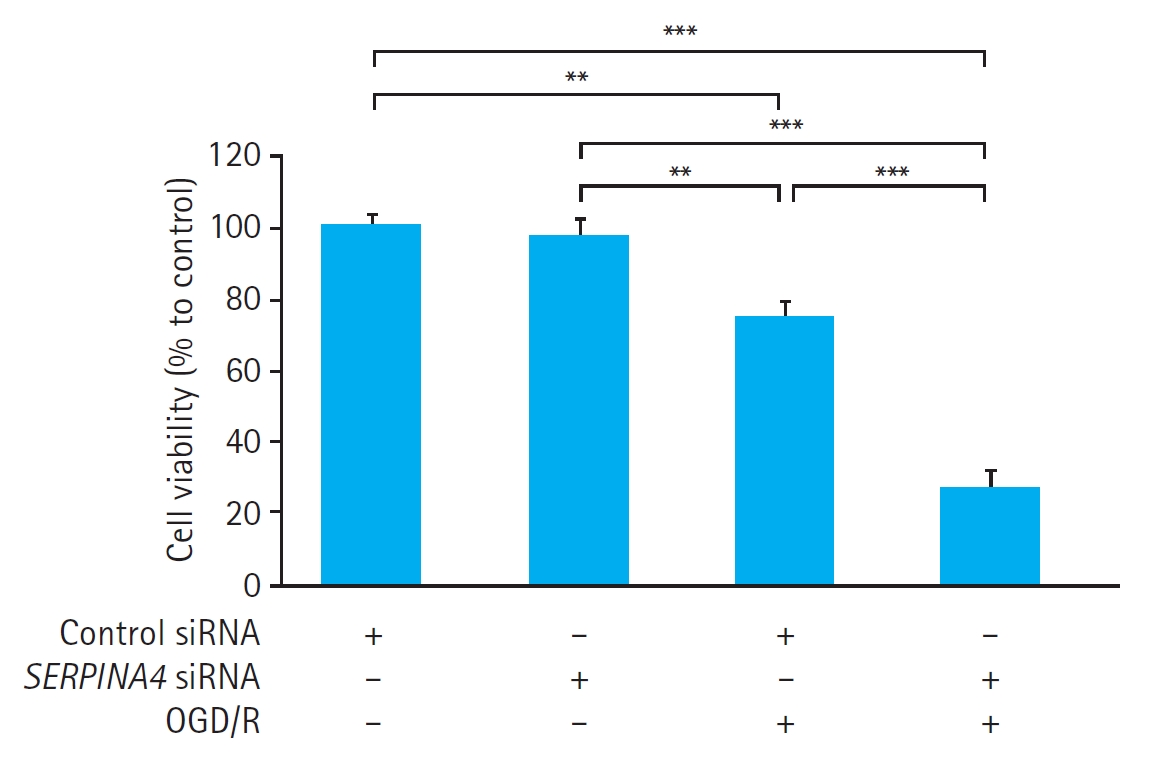
Effects of SERPINA4 knockdown on cell viability and kallistatin expression in HUVECs exposed to OGD/R
In HUVECs transfected with either control siRNA or SERPINA4 siRNA, OGD/R significantly reduced cell viability (control siRNA, P<0.01; SERPINA4 siRNA, P<0.001) (Fig. 3). Cell viability in HUVECs exposed to OGD/R was significantly lower in the SERPINA4 knockdown cells compared with HUVECs transfected with control siRNA (P<0.001).
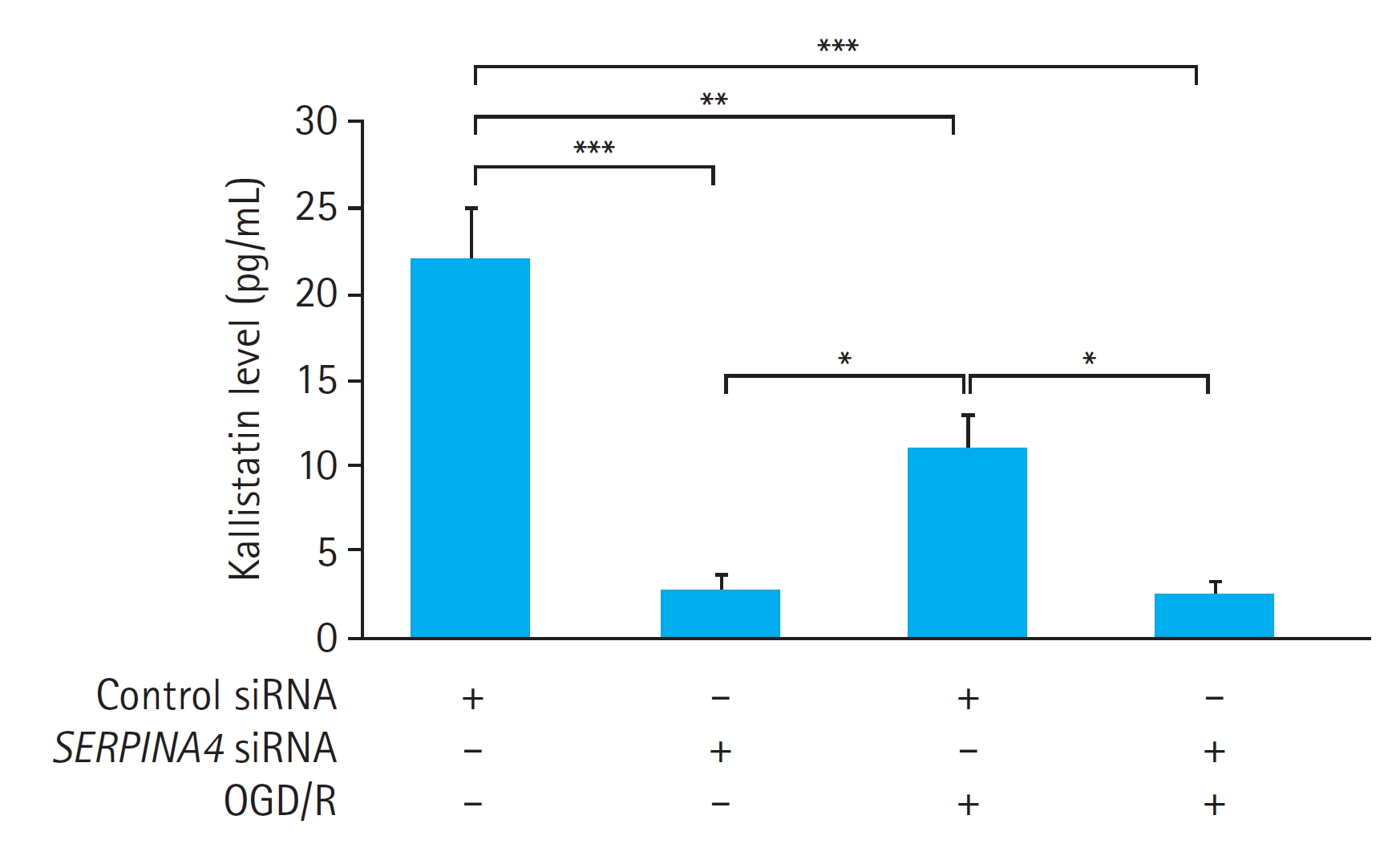
We also measured the kallistatin concentration in the culture medium of the HUVECs with or without OGD/R. OGD/R significantly reduced the kallistatin concentration in the culture medium of the HUVECs transfected with control siRNA (P<0.01). However, OGD/R did not induce a significant difference in the kallistatin concentration in the culture medium in HUVECs with SERPINA4 knockdown (P=0.999). SERPINA4 knockdown led to a significant reduction in kallistatin concentration regardless of OGD/R, with a more pronounced effect observed without OGD/R (without OGD/R, P<0.001; with OGD/R, P<0.01) (Fig. 4).
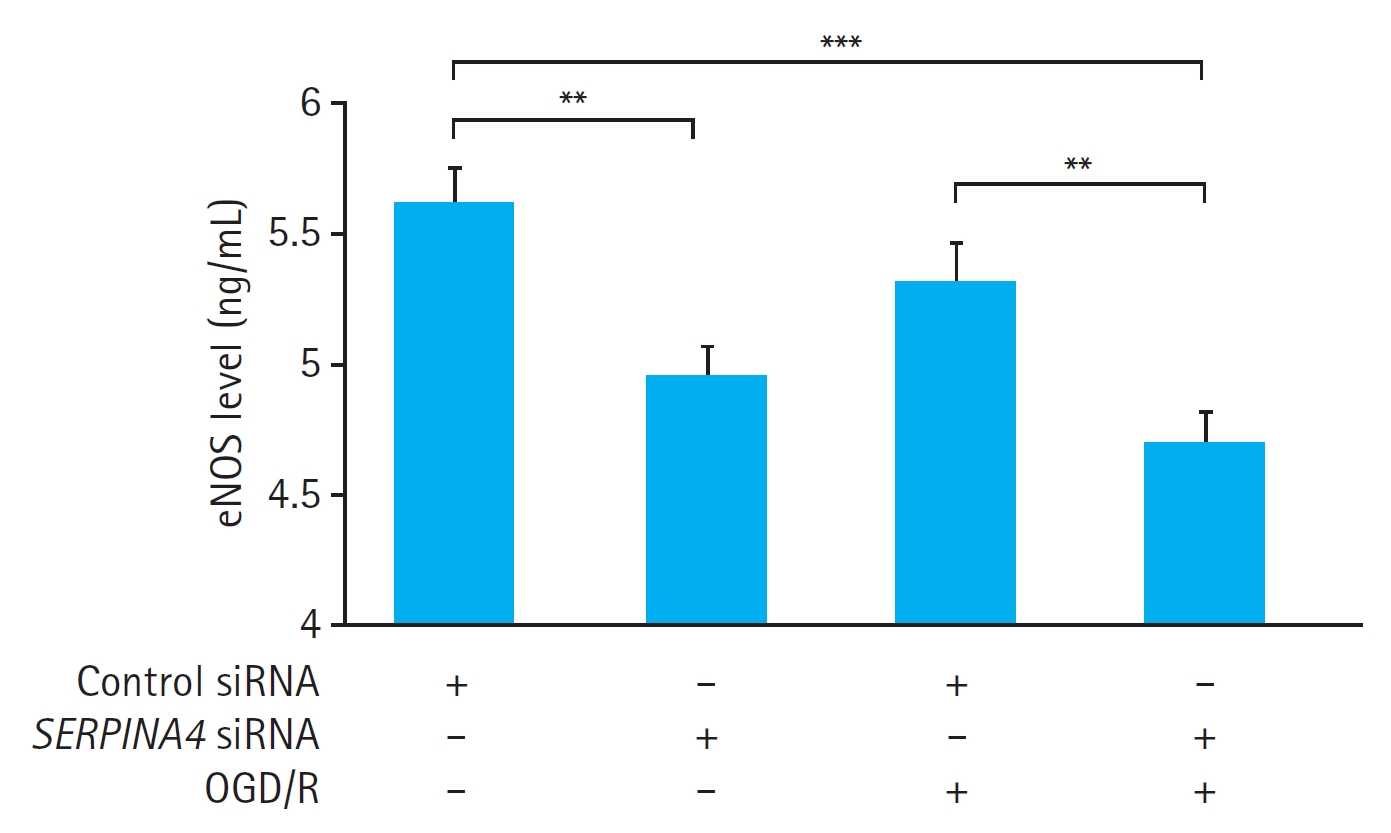
Effects of SERPINA4 knockdown on eNOS expression and total NO concentration in HUVECs exposed to OGD/R
OGD/R did not induce a significant difference in the eNOS concentration in the culture medium of the cells transfected with either control siRNA (P=0.362) or SERPINA4 siRNA (P=0.527) (Fig. 5). Nevertheless, SERPINA4 knockdown resulted in a significant reduction in the eNOS concentration in the culture medium regardless of OGD/R (P<0.01).
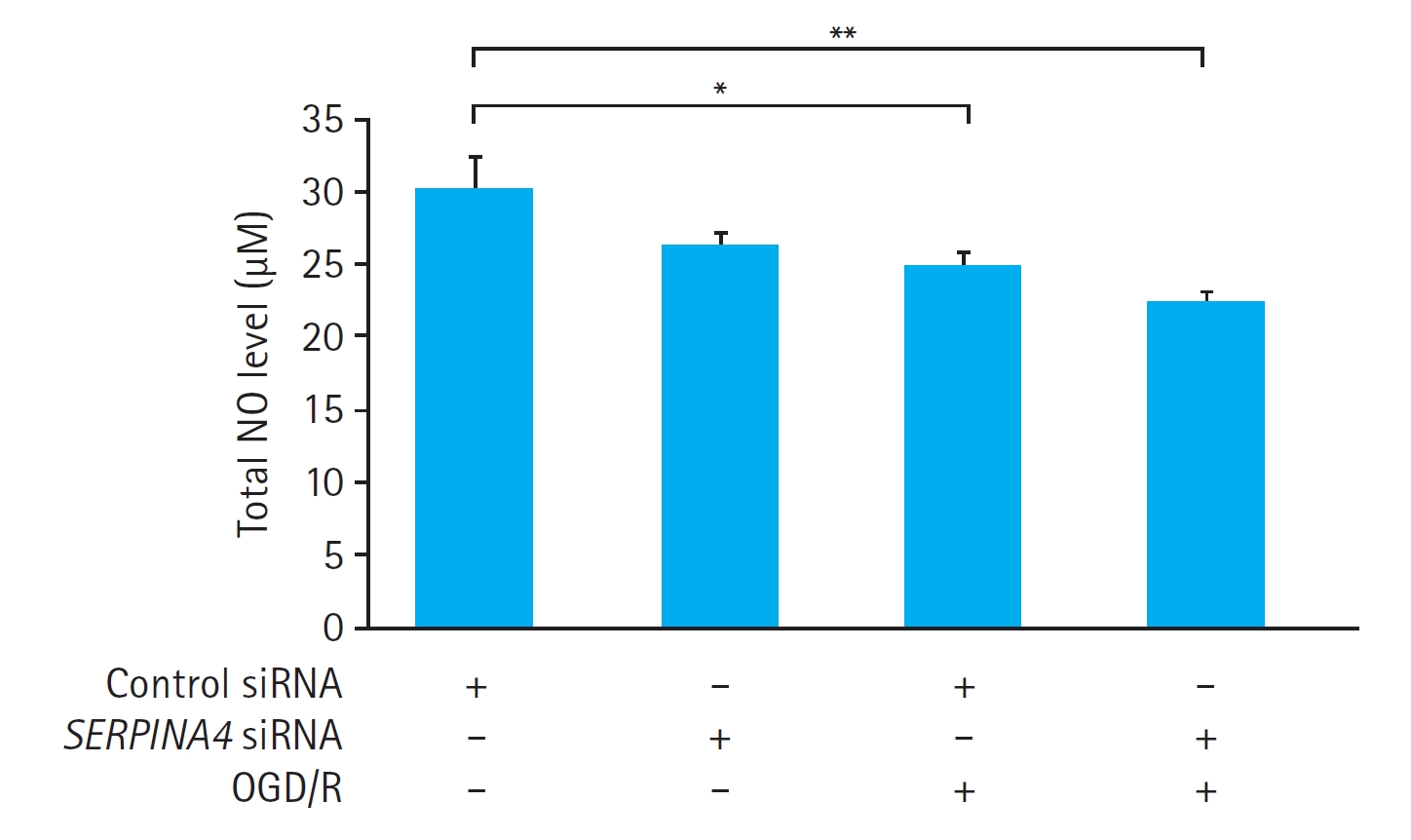
OGD/R significantly reduced the total NO concentration in the culture medium of the HUVECs transfected with control siRNA (P<0.05). In contrast, OGD/R did not induce a significant change in the total NO concentration in the culture medium of the SERPINA4 knockdown cells (P=0.17). Additionally, the total NO concentration in the culture medium of HUVECs exposed to OGD/R was not significantly different between the HUVECs transfected with control siRNA and SERPINA4 knockdown cells (P = 0.728) (Fig. 6).
DISCUSSION
In the present study, we investigated the antioxidant mechanism of kallistatin in preventing IR injury. We found that exposure to OGD/R reduced HUVEC viability, and this effect was more pronounced in the SERPINA4 knockdown cells. SERPINA4 knockdown significantly decreased eNOS concentrations induced by OGD/R (P<0.01) but did not significantly affect the change in total NO concentration (P=0.728). These results confirm the protective role of kallistatin against IR injury, implying that its potential antioxidant mechanism might be attributed to its enhancement of eNOS expression.
Our study provides experimental evidence demonstrating that a deficiency of kallistatin led to reduced cell viability in response to IR injury. We used the OGD/R method to mimic IR injury. OGD/R is commonly used as an in vitro model for simulating ischemic stroke, and it has been widely accepted as a representative model that mirrors the conditions observed in in vivo models of brain ischemia [18,19]. Additionally, OGD-induced cardiac myocyte injury is commonly used as a model for myocardial IR injury during cardiovascular disease [20ŌĆō22]. OGD/R is also used as an acute kidney injury model to replicate the effects of renal IR [23]. Given the extensive use of OGD/R in previous studies to investigate IR injury, including studies involving HUVECs [24ŌĆō26], we selected this model to replicate IR injury in HUVECs for our study.
A previous study [12] demonstrated that kallistatin enhanced eNOS levels and NO production in a dose-dependent manner, which was mediated by kallistatin binding protein Kruppel-like factor 4. Although it was the first study that highlighted the eNOS-promoting effect of kallistatin, this study primarily aimed to investigate the anti-inflammatory effect of kallistatin rather than the antioxidant mechanism. Additionally, the study did not explore the impact of kallistatin administration on cell survival. Another study that investigated the protective role of kallistatin against oxidative stressŌĆōinduced endothelial cell injury revealed that kallistatin suppressed tumor necrosis factor-╬▒ (TNF-╬▒)-induced ROS formation and cellular apoptosis [13]. The effect was blocked by the knockdown of eNOS expression. Although Shen et al. [13] suggested that eNOS mediated the cell-protective effect of kallistatin against oxidative stress, it did not explore the direct protective effect of kallistatin against IR injury by kallistatin depletion or knockdown. Several studies have used kallistatin knockdown techniques [27ŌĆō29]. One study [28] used kallistatin knockdown mice to investigate the role of kallistatin in endothelial senescence. The results confirmed that kallistatin knockdown exacerbated lung endothelial cell senescence and resulted in reduced levels of eNOS. However, the study did not use an IR injury model and primarily focused on senescence without specifically investigating cell viability. Our study is distinct with previous research in several ways. First, we used the OGD/R model to induce IR injury in HUVECs, with the primary objective of investigating the antioxidant mechanism. Second, we used kallistatin knockdown cells and assessed cell viability to examine the direct cell-protective effect of kallistatin, not the effect of its mediator eNOS or NO. However, we also measured eNOS and NO expression to elucidate kallistatin's antioxidative mechanism, particularly through its eNOS-promoting effect.
This study has several limitations. First, we were unable to directly confirm whether the suppression of kallistatin expression had a statistically significant effect on the alteration of NO expression in HUVECs exposed to OGD/R. Kallistatin promotes NO production by enhancing the expression of eNOS. NO, in turn, exhibits antioxidative and anti-inflammatory effects by inactivating various proteins such as vascular endothelial growth factor, TNF-╬▒, transforming growth factor-╬▓, nuclear factorŌĆō╬║B, and NADPH oxidase [11]. Therefore, it is crucial to confirm the direct link between kallistatinŌĆÖs eNOS-promoting effect and increased NO production to explain its antioxidant property. However, this link was not confirmed in our study. Notably, although not statistically significant, the mean total NO concentration of HUVECs exposed to OGD/R was lower in SERPINA knockdown cells (22.3 ╬╝M) compared with HUVECs transfected with control siRNA (24.6 ╬╝M), suggesting that eNOS suppression from kallistatin knockdown might have impacted NO concentration. Thus, further investigation to fully elucidate the relationship between kallistatin, eNOS, and NO production in the context of IR injury is warranted.
Second, our study focused solely on investigating the protective effect of kallistatin against IR injury primarily attributed to its antioxidative property. Kallistatin possesses diverse biological activities, in addition to its antioxidative function, including anti-inflammatory and anticoagulant properties [10]. While oxidative stress is a critical factor contributing to tissue injury by IR processes, tissue hypoperfusion induced by local inflammation and coagulation also plays a significant role [30]. Considering the multifaceted nature of kallistatin functions, it is likely that its protective effect against IR injury arises from a combination of its antioxidative, anti-inflammatory, and anticoagulant properties. Thus, the precise mechanisms underlying kallistatin's protective effects need further investigation, which will contribute to the comprehensive understanding of its various functions. Additionally, assessing also the morphological changes of the cells would contribute to a deeper understanding of the underlying mechanisms.
Third, this study focused on in vitro experiments. Consequently, we are unable to definitively conclude that kallistatin suppression induces susceptibility to IR injury in an in vivo environment. To overcome these limitations and to determine the protective effect of kallistatin on IR injury in vivo, animal experiments with the inhibition of kallistatin expression on nerve cells are warranted.
Fourth, this study was based on our previous research [17], which primarily focused its clinical attention on cardiac arrest patients. However, the in vitro model used in this study was not the model representing cardiac arrest. To date, knowledge regarding the mechanism of action of kallistatin in cardiac arrest patients is limited. Tissue ischemia caused by cardiac arrest induces hemodynamic and metabolic disturbances, leading to tissue injury [30ŌĆō32]. Subsequent reperfusion through cardiopulmonary resuscitation (CPR) and the return of spontaneous circulation results in the generation of ROS, leading to further oxidative cell injury with compromised cell membrane and mitochondrial function [30ŌĆō32]. Consequently, it can be inferred that cardiac arrest and subsequent CPR represent typical forms of IR injury, suggesting that kallistatin may play a role in this process. Therefore, to further investigate the role of kallistatin in the context of cardiac arrest, future research using cardiac arrest models in small animals or in vitro models using myocyte cells will be necessary.
Finally, an experiment to determine the effect of kallistatin administration on IR injury was not conducted. To become a therapeutic target for improving outcomes, a substance must be associated with a poor prognosis, and addition of the substance should improve the outcome [33,34]. However, in this study, the preparation of an adenovirus vector containing human kallistatin and the purification of recombinant kallistatin were too complicated, and it was difficult to determine the timing of kallistatin administration in the OGD/R model. Thus, experiments to confirm the effect of kallistatin administration were not conducted. To ascertain the potential therapeutic benefits of kallistatin, further studies investigating the effects of kallistatin supplementation on IR injury are warranted. In one study [15] that investigated the effect of kallistatin on cardiac function after myocardial IR injury, kallistatin gene delivery significantly reduced IR-induced cardiomyocyte apoptosis, promoted cardiac eNOS activation, and increased NO formation. However, the IR model used in the previous study [15] involved ligation of the left anterior descending coronary artery, which is not representative of cardiac arrest. Therefore, if cell or animal studies with a cardiac arrest model validate that kallistatin supplementation yields a protective effect against IR injury, it could provide valuable insights into the potential utilization of kallistatin as a therapeutic target.
In summary, transfection of HUVECs with SERPINA4 siRNA effectively suppressed kallistatin expression. Knockdown of SERPINA4 resulted in reduced cell viability in response to OGD/R and significantly affected the change in eNOS level induced by OGD/R. These findings suggest that the protective role of kallistatin against IR injury might contribute to its property of enhancing eNOS expression. Further animal studies are needed to confirm the protective effect of kallistatin against IR injury in an in vivo environment.